1Laboratory of Genetics and Genomics, Institute for Developing Science and Health Initiatives, Dhaka, Bangladesh; 2Department of Biochemistry and Molecular Biology, University of Dhaka, Dhaka, Bangladesh; 3Infectious Disease Laboratory, Institute for Developing Science and Health Initiatives, Dhaka, Bangladesh;
![]() 10.3329/bmrcb.v45i3.44642
10.3329/bmrcb.v45i3.44642 ![]() 0000-0002-2394-0447
0000-0002-2394-0447
Background: Thalassaemia is one of the most common genetic blood disorders worldwide. Patients with β-thalassaemia major and HbE/β-thalassaemia are blood transfusion dependent. Foetal haemoglobin or HbF can play a role in disease manifestations in these patients and there is evidence that a homozygous state for XmnI polymorphic site, associated with increased expression of Gγ-gene, may play an important role among other factors in ameliorating the clinical severity of homozygous β-thalassaemia and thalassaemia intermedia. The aim of this review was to provide a comprehensive review of the role of XmnI polymorphic site for increased HbF production in HbE/β and β-thalassaemia patients
Methods Published literatures were reviewed on the allelic frequency of Xmn1 polymorphism and its effect on HbF induction among thalassaemia patients of different countries.
Results: In all β-thalassaemias, Hb F levels are relatively increased due to the selective survival of the erythroid precursors that synthesize relatively more γ-chains. The expression of HbF level is dominated by three different loci: HBG2: γ -158C>T, BCL11A, and HBS1L-MYB intergenic region. Genetic determinants influencing Hb F response can be within the β-globin complex or trans-acting. The published literature showed that the C>T substitution (rs7482144) at position –158 of the Gγ-globin gene, referred to as the XmnI-Gγ polymorphism, is a common sequence variant in all population groups, present at a frequency of 0.32 to 0.35. It was found in some studies, response to Hydroxyurea (HU) has been shown to be largely associated with the presence of the C>T polymorphism at -158 XmnI site (HBG2:c.- 53-158C>T) upstream of the Gγ-globin gene and HU therapy exerts a 2- to 9- fold increase in γ-mRNA expression in β-thalassaemia patients.
Conclusions: A number of various study groups around the world suggests that XmnI polymorphism is an important key regulator of disease severity of HbE/β and β-thalassaemia patients.
Keywords: β-thalassaemia, HbE/β-thalassaemia, XmnI-Gγpolymorphism, Hydroxyurea.The word “thalassaemia” derived from the Greek words “Thalassa” (sea) and “Haema” (blood) refers to the disorders associated with defective synthesis of α- or β-globin subunits of haemoglobin (Hb) A (α2β2). The diseases are inherited as pathologic alleles of one or more of the globin genes located on chromosomes 11 (β) and 16 (α).1 The thalassaemia syndrome is characterized based on the affected globin chains. Alpha (α) thalassaemia is caused by reduced (α+) or absent (α°) synthesis of alpha globin chains, and beta (β) thalassaemia is caused by reduced (β+) or absent (β°) synthesis of beta globin chains. 1, 2 More than 200 deletions or point mutations that impair transcription, processing, or translation of α- or β-globin mRNA have been identified.1,3,4,9 However, only 20 mutations account for 90% of the abnormal β-genes.5 Haemoglobin E (HbE) is another abnormal structural variant of haemoglobin, resulting from a substitution mutation G>A in codon 26 (Glu>Lys) of the β-globin gene, mostly prevalent in South-East Asian populations.6,7,8,10 HbE/β-thalassaemia results from co-inheritance of a β-thalassaemia allele from one parent and the structural variant Haemoglobin E from the other.7,8,11. Worldwide, HbE/β-thalassaemia may be one of the most important haemoglobinopathies because of the high gene frequencies for both HbE and β-thalassaemia.12-14
Thalassaemia is one of the most common genetic blood disorders worldwide.15-17 It is estimated that more than 300,000 infants are born with major haemoglobinopathies worldwide each year of whom 60,000 to 70,000 are β-thalassaemia major cases especially in the Mediterranean area, Middle East, Far East, and East Asia.18-20 Globally every year, severe form of β-thalassaemia accounts for 50,000 to 100,000 deaths in all age group and about 0.5%- 0.9% deaths of under-5 children of low or middle income countries.21 According to Thalassaemia International Federation (TIF), about 23,000 children are born with transfusion-dependent β-thalassaemia major each year, while a smaller ill-defined number have the non-transfusion dependent thalassaemia (NTDT), a form of β-thalassaemia intermedia.22 Bangladesh, the most densely populated countries in the world, with a population of over 160 million people. According to World Health Organization (WHO), approximately 3% of the carriers of β-thalassaemia and 4% are the carriers of haemoglobin E (HbE) in Bangladeshi population.23 It is highly concerning that with the birth rate of 21.6/1000, it could be estimated that nearly 2500 thalassaemia major cases are added every year in Bangladesh.24 As, thalassaemia is a hereditary disease, it is only manageable when it is prevented. So, proper and effective public awareness should be built up among the population of Bangladesh.
However, the thalassaemias are heterogeneous at the molecular level, with more than 200 disease causing mutations having been identified.12 In erythroid development, the γ-globin expression was regulated by interactions between cis-acting sequences within the β-globin cluster and trans-acting factors such as BCL-11A, cMYB, and TOX.12,25 The expression of HbF level is dominated by three different loci:HBG2: γ -158C>T on 11p15.4, BCL11A on 2p16.1 and HBS1L-MYB intergenic region on 6q23.3. The most significant genetic factor in cis associated with high HbF is XmnI polymorphism located at -158 upstream to the Gγ-globin genes.26 Although the production of Hb F is almost switched off at birth, all adults continue to produce residual amounts of Hb F. In all β-thalassaemias, HbF levels are relatively increased due to the selective survival of the erythroid precursors that synthesize relatively more γ-chains.4 Xmn1 polymorphism is responsible for the induction of γ-chains in adult patients with β-thalassaemias.
A literature review was performed with the aim to probe the role of XmnI-Gγ polymorphism on HbF induction in thalassaemia patients. The articles were searched at PubMed, google scholar and other Journal databases. The key words such as thalassaemia, β-thalassaemia, HbE/β-thalassaemia, XmnI-Gγ polymorphism, Haemoglobin F and Hydroxyurea were used while searching these sources. Primarily, the articles which addressed the modifying effect of XmnI-Gγ polymorphism on disease severity of the HbE/β and β-thalassaemia patients were screened. Finally, a total of 81 articles, both original and review, were selected for this review purpose. Articles not written in English were excluded.
Pathophysiology and Clinical Variability of β-thalassaemia and HbE/ β-thalassaemia: Although clinical spectra vary depending on coinheritance of other genetic modifiers, the underlying pathology among the types of thalassaemia is similar.27 This pathology is characterised by decreased Hb production and red blood cell (RBC) survival, resulting from the excess of unaffected globin chain, which form unstable homotetramers that precipitate as inclusion bodies. α-homotetramers in β-thalassaemia are more unstable than β-homotetramers in α-thalassaemia and therefore precipitate earlier in the RBC life span, causing marked RBC damage and severe haemolysis associated with ineffective erythropoiesis (IE) and extramedullary hemolysis.28 (figure:1) Without transfusion support, 85% of patients with severe homozygous or compound heterozygous β-thalassaemia will die by 5 years of age because of severe anaemia.29
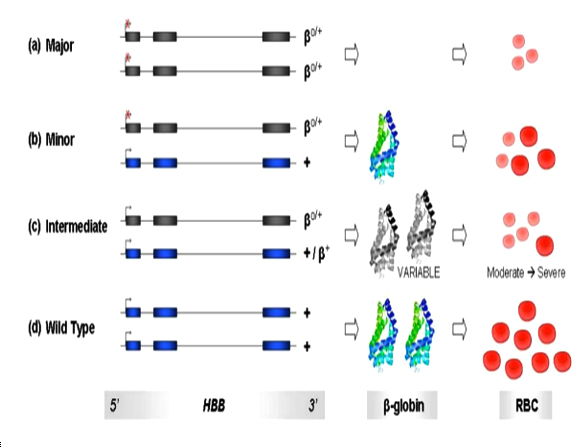
β-thalassaemia includes three main forms: Thalassaemia Major, variably referred to as "Cooley's Anaemia" and "Mediterranean Anaemia", Thalassaemia Intermedia and Thalassaemia Minor also called "β-thalassaemia carrier", "β-thalassaemia trait" or "heterozygous β-thalassaemia". Individuals with β-thalassaemia major (β0/β0, β0/β+, and sometimes β+/ β+) usually come to medical attention within the first two years of life and require regular RBC transfusions to survive.9 (figure 2a) Patients with β-thalassaemia intermedia (β+/β0 or, β+/ β+) have milder anemia and do not require or only occasionally require transfusion.9(figure 2c).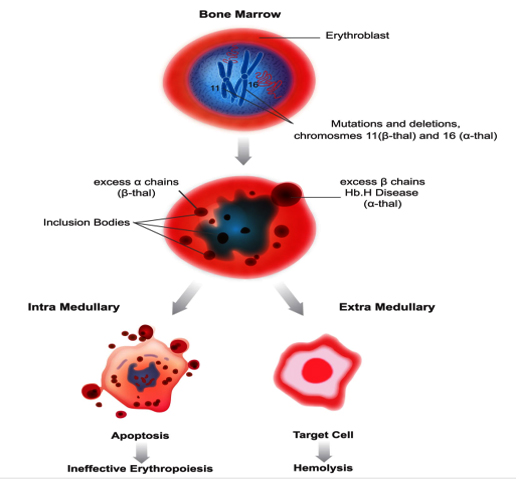
And individuals with β-thalassaemia minor (β/β+, β/β0 or mild β/β+) are carriers and they are usually clinically asymptomatic but sometimes have a mild anemia. Table-1 illustrates common genotypes leading to a β-thalassaemia intermedia phenotype.30 (figure 2b)
The HBB variants are represented in grey exons while the wild type alleles are represented in blue exons. Production of β-globin from a single/double wild type alleles are represented by one/two colored schematic of the β-globin protein respectively. Grey colored β-globin diagrams refer to below-normal synthesis levels of the protein, created by mutant HBB variants. Bright red-colored RBCs represent normal cell phenotype, while pink colored ones represent microcytic, hypochromic cells characteristic of beta-thalassaemia phenotype. Relative number of RBC reflects relative levels of anaemia amongst the three classes of β-thalassaemia and in comparison to the wild type RBC pool.
Phenotype |
Genotype |
Clinical severity |
Silent carrier |
|
|
Trait/minor |
|
|
Intermedia |
|
|
Major |
|
|
Hb E/β-thalassaemia results from co-inheritance of a β-thalassaemia allele from one parent and the Haemoglobin E, structural variant of haemoglobin, from the other. Haemoglobin E results from a G>A substitution in codon 26 of the β globin gene, which produces a structurally abnormal Haemoglobin (HbE).31 The pathophysiology of Hb E/β-thalassaemia is related to many factors including reduced β-chain synthesis resulting in globin chain imbalance, ineffective erythropoiesis, apoptosis, oxidative damage and shortened red cell survival.32,33 Depending on the severity of symptoms, HbE/β-thalassaemia can be divided into three categories.9 Individuals with mild HbE/β-thalassaemiamaintain Hb levels between 9 and 12 g/dl and usually does not develop clinically significant problems. Individuals with moderately severe HbE/β-thalassaemia maintain Hb levels between 6 and 7 g/dl and the clinical symptoms are similar to thalassaemia intermedia. The Hb level can be as low as 4-5 g/dl in the patients with severe HbE/β-thalassaemiausually manifest symptoms similar to thalassaemia major and are treated as thalassaemia major patients.
The Globin Genes: There are eight functional globin genes as well as several pseudo genes. The globin genes are found in two loci, each of which has an associated upstream regulatory element. The α-globin locus on chromosome 16 34,35 contains three of globin genes. Listed in 5’ to 3’ order these are Haemoglobin subunit zeta (HBZ), Haemoglobin subunit alpha 2 (HBA2) and Haemoglobin subunit alpha 1 (HBA1)36 (figure:3)
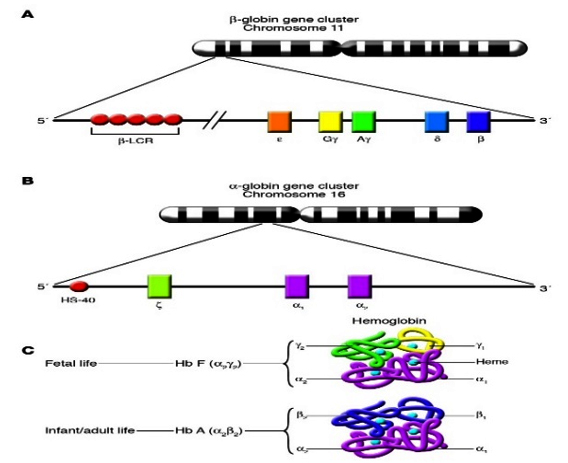
The genes of the β-globin gene cluster (ε, Gγ, Aγ, δ, and β) are present on chromosome 11 in the same order in which they are expressed during development. The β–locus control region (β–LCR) is a major regulatory element located far upstream of the genes of the cluster that is necessary for the high level of expression of those genes. (B) The genes of the α-globin gene cluster (ζ, α1, and α2) are present on chromosome 16, also in the same order in which they are expressed during development. HS-40 is a major regulatory element located far upstream of the genes of the cluster that is necessary for their high level of expression. (C) During fetal life, Hb F (α2γ2) is the predominant type of Haemoglobin. Haemoglobin switching refers to the developmental process that leads to the silencing of γ-globin gene expression and the reciprocal activation of adult β-globin gene expression. This results in the replacement of Hb F by Hb A (α2β2) as the predominant type of Haemoglobin in adult life. (Frenette PS and Atweh GF. 2007)
The remaining five functional globin genes are found in the β-globin locus on chromosome 11.37,38 Listed in 5’ to 3’ order these are Haemoglobin subunit epsilon 1 (HBE1), Haemoglobin subunit gamma 2 (HBG2), Haemoglobin subunit gamma 1 ((HBG1), Haemoglobin subunit delta (HBD) and Haemoglobin subunit beta (HBB). An upstream regulatory element known as the β-Locus Control Region (β-LCR) is required for expression of these genes.39 Fetal Hb (HbF, α2γ2) is the predominant form during fetal development but is largely replaced by adult Hb (HbA, α2β2) following a shift from gamma (γ)- to β-globin gene expression that begins around birth.40 Two main mechanisms control globin gene switching, competition for access to the upstream regulatory element and autonomous gene silencing.41 Autonomous gene silencing plays an important role in the switching from foetal to adult Haemoglobin.
Haemoglobin F: Haemoglobin F (HbF, α2γ2) accounts for up to 90% of the circulating Haemoglobin at birth. It’s synthesis starts to decline during the third trimester, and over the first year of life itis gradually replaced by adult Haemoglobin, HbA (α2β2). Normal adults have less than 1% of HbF, apparently confined to a subsetof red blood cells called F cells,42 which constitute about 3% of the erythrocytes.43 Several inherited and acquired conditions are associated with the persistence or the reactivation of HbF production.44Most of the genetic disorders associated with persistent HbF production involve alterations of the structure of the β-globin cluster. The highest adult levels of HbF are seen in β- and δβ-thalassaemia, or hereditary persistence of foetal haemoglobin (HPFH), in which HbF can constitute up to 100% of the Haemoglobin. It is now clear that HPFH is an extremely heterogeneous group of conditions, some of which result from deletions of the β-globin gene or point mutations in the γ-globin gene promoter regions, whereas others arise from genetic determinants that segregate independently of the β-globin gene cluster.45
Several acquired conditions are associated with modest elevations of HbF. They include pregnancy, recovery from marrow hypoplasia, aplastic anaemia, leukemia, thyrotoxicosis, haepatoma, and juvenile chronic myeloid leukaemia.46 The latter condition is exceptional in that it seems to reflect a genuine reversion to fetal erythropoiesis.47 The remainder seem to be examples of the transient reactivation of HbF under conditions of acute erythropoietic stress, that is, rapid expansion of theerythron.48
Mechanism for increased HbF production: According to Rees DC et al, a proposed possible mechanism by which HbF may be increased.44 In this model, the absolute numbers of F-cell progenitors would expand, proportionate to the increase in all red blood cell precursors. In both transfused and non-transfused patients, the F-cell precursors would have a selective advantage because of their lesser degree of globin chain imbalance, leading to the observed increases in HbF levels. The observed change in α/δ ratio is not compatible with this mechanism alone, and suggests the possibility that there is a genuine increase in HbF and/or F-cell production. This preferential production of F reticulocytes has typically been thought to be important in acute increases in Epo, rather than the chronic elevations seen in thalassaemia figure 4.48,49
Association of XmnI polymorphism with Hb F induction: Genetic determinants influencing Hb F response can be within the β-globin complex or trans-acting.

The C>T substitution (rs7482144) at position –158 of the Gγ-globin gene, referred to as the XmnI-Gγ polymorphism, is a common sequence variant in all population groups, present at a frequency of 0.32 to 0.35.50,51 Although the increases in Hb F and F cells are minimal in normal people, clinical studies have shown that, under conditions of hematopoietic stress, for example in homozygous β-thalassaemia and sickle cell disease, the presence of the Xmn1-Gγsite favors a higher Hb F response.52,53 However, the -158 C > T polymorphism is located near a nuclease hypersensitive site at 50 to 150 bp upstream region of the Gγ-globin gene. Perhaps the -158 substitution reduces the binding of transcription factor(s) that silence(s) the γ-globin gene expression in adult cells. Therefore, the γ-globin gene is reactivated in adult life.54,55
The Hb F is a mixture of two molecular species α2Gγ2 and α2Aγ2 in which the constituent γ-chains contain a glycine or an alanine at position 136. During the switch from fetal to adult, there is a quantitative change in the γ-chain composition. Normally the Gγ: Aγ ratio is 70:30 at the time of birth and 40:60 in the trace amounts of Hb F found in the adult. This ratio is modified in many Haemoglobin disorders, but in the presence of XmnIpolymorphic site almost this ratio look like the time of birth.56
The frequency of XmnI polymorphic site: The prevalence of XmnIpolymorphic site 5' to the Gγ-gene is different among various population (table II). There is evidence that a homozygous state for Xmn1 polymorphic site, which is associated with increased expression of Gγ-gene, may play an important role among other factors in ameliorating the clinical features of homozygous β-thalassaemia and its clinical presentation as thalassaemia intermedia.57 Furthermore, the presence of XmnIpolymorphic site 5' to the Gγ-globin promoter region was positively correlated with elevated synthesis of fetal Hb and its Gγ-globin component in term newborn infants and is associated with delayed switch over from fetal to adult Haemoglobin. It is unknown that how XmnIpolymorphic site influences the expression of the Gγ-globin gene. It seems that interaction of a multi-protein transcription complex to be involved. In a genome-wide linkage study of large Asian Indian kindred, a genetic interaction between the XmnI polymorphic site and a locus on chromosome 8q was reported to influence on adult F cell (FC) levels.58,59 Unlike the rare mutations in the γ-globin promoter that are consistently associated with large discrete effects of increased HbF levels of 10–35% in heterozygotes, the so-called pancellular hereditary persistence of fetal Haemoglobin (HPFH), the change at Gγ -158 does not always raise the Hb F levels in otherwise normal individuals. The XmnI polymorphic site is not a recognized binding motif for any of the known transcription factors.60
However, the Hb F response associated with the XmnI polymorphic site is usually moderate and may not be sufficient to explain the wide difference in phenotype observed in some cases.61,62 According to Ballas SK et al.63 there was a significant correlation between the presence of Xmn1 polymorphic site and increased Gγ: Aγ ratio. However, the Hb F level was not significantly increased in the presence of Xmn1 polymorphic site in their study. Although XmnI polymorphic site maintains a Gγ: Aγ ratio typical of fetal life but does not necessarily cause elevation of Hb F. The latter seems to depend on factors other than the XmnI polymorphic site.63 Hooshang N et al revealed that in the presence of XmnIpolymorphic site Gγ percent and Gγ: Aγratio were significantly increased (p=0.01) and the clinical features such as splenomegaly and bone marrow expansion were significantly improved (p=0.01). It was found that in the presence of Xmn1polymorphic site on both chromosomes (+/+ the level of Hb F tended to be increased compared to the absence of Xmn1(-/-) (table III).60
Country/Population groups |
Sample Size (n) |
Types of population |
Frequency |
References no. |
Caucasian French |
100 |
Normal infants |
0.32 |
56 |
European |
300 |
Healthy |
0.32–0.35 |
50 |
India |
64 |
β-thalassaemia |
0.25 |
77 |
Eastern India |
64 |
β-thalassaemia and HbE/ |
0.48 |
78 |
Northern India |
101 |
β-thalassaemia major and intermedia |
0.28 |
79 |
Southern Iran |
48 |
β-thalassaemia intermedia |
0.41 |
80 |
Western Iran |
197 |
β-thalassaemia major |
0.39 |
60 |
Malaysia |
107 |
β-thalassaemia major |
0.66 |
81 |
Parameter |
Xmn1 C/C (+/+) |
Xmn1 C/T (+/-) |
Xmn1 T/T |
p value |
Hb F level % |
97.1 |
94.4 |
91.8 |
0.08 |
Gγ % |
74.8 |
71.4 |
66.7 |
0.01 |
Aγ % |
25.1 |
28.6 |
33.3 |
0.01 |
Gγ/Aγ ratio |
3 ± 0.5 |
2.5 ± 0.4 |
2 ± 0.3 |
0.01 |
Age of first blood transfusion (months) |
12 ± 8 |
11 ± 5 |
10 ± 5 |
0.16 |
Facial bone deformity % |
17.5 |
25.4 |
57.1 |
0.02 |
Splenectomy % |
15.9 |
19.1 |
65 |
0.01 |
Gγ-XmnI polymorphism genotyping: XmnI polymorphism is heterogeneously distributed in different parts of the world.64 The archived genomic DNAs are genotyped employing Polymerase Chain Reaction Restriction Fragment Length Polymorphism (PCR-RFLP) technique. The genotypes were categorised into homozygous wild type (CC, +/+), heterozygous (CT, +/-) and homozygous variant (TT,-/-)60,65-68 Another study was performed to detect the Gγ-XmnI polymorphism genotype in patients using the Tetra-Primer ARMS-PCR technique.69
Association of Gγ-XmnI polymorphism with Hydroxyurea (HU) treatment:HU therapy has been successfully used in thalassaemia intermedia patients and was associated with a signifcant improvement in hematological parameters and quality of life.70,71Hydroxyurea efficacy in β-thalassaemia major has been variable in different studies.72-74 Response to HU has been shown to be largely associated with the presence of the C>T polymorphism at -158 Xmn1 site (HBG2:c.- 53-158C>T) upstream of the Gγ-globin gene and it is thus far the most studied nucleotide change to have a significant association to drug response. This particular polymorphism acts as an enhancer of HbF expression during erythropoietic stress, resulting in a beneficial effect in SCD patients. HU is a myelo suppressive agent that may enhance fetal Haemoglobin production. Several pharmacologic agents, such as 5-azacytidine, erythropoietin, butyrates including Hydroxyurea have been shown to stimulate γ-globin gene expression in vivo and therefore might reduce the severity of clinical symptoms in patients with intermediate thalassaemia. Moreover, one study on β-thalassaemia patients treated with Hydroxyurea has revealed a significant correlation between the presence of T allele in Xmn1 polymorphic site and the better treatment response. Hydroxyurea therapy exerts a 2- to 9- fold increase in γ-mRNA expression in β-thalassaemia patients,74leading to improvement in the a/non–a-chain imbalance andmore-effective erythropoiesis.75However, Kosaryan et al. suggested that β-thalassaemia major or intermedia with Xmnl polymorphism of ( C/T or +/–) show better response to hydroxyurea therapy than ( T/T or –/–) genotype, this finding was not proved by other studies.76No previous study on association of Gγ-XmnI polymorphism with Hydroxyurea (HU) treatment in Bangladeshi populationis found.
Conclusion
The high level of HbF can ameliorate the severity of the disease by reducing the excess alpha chain imbalance in patients with Sickle cell disease (SCD), β-thalassaemia and HbE/β-thalassaemia. Therefore, for investigating the success of Hydroxyurea medication in diseased population, a follow-up study is required to determine the HbF induction effect of Hydroxyurea in presence of T allele in thalassaemia patients. However, till date there is no study regarding the frequency of XmnI-Gγ polymorphism and its effect on HbF production among thalassaemia patients in Bangladesh. So, study on the effect of XmnI-Gγ polymorphism on disease severity of Bangladeshi thalassaemia patients is recommended in order to validate the use of Hydroxyurea as therapeutic intervention for these patients.
References
- Rachmilewitz EA, Giardina PJ. How I treat thalassaemia. Blood. 2011; 118: 3479-88.
- Muncie JH, Campbell J. Alpha and beta thalassaemia. American family physician. 2009; 80:339-44.
- Ma Q, Abel K, Sripichai O, Whitacre J, Angkachatchai V, Makarasara W, et. al. β‐Globin gene cluster polymorphisms are strongly associated with severity of HbE/β0‐thalassaemia. Clinical genetics. 2007; 72:497-505.
- Thein SL. Genetic modifiers of beta-thalassaemia. Haematologica. 2005; 90:649-60.
- Kazazian HJ, Boehm CD. Molecular basis and prenatal diagnosis of beta-thalassaemia. Blood 1988; 72:1107-16.
- Ma Q, Abel K, Sripichai O, Whitacre J, Angkachatchai V, Makarasara W, Winichagoon P, Fucharoen S, Braun A, Farrer LA. β‐Globin gene cluster polymorphisms are strongly associated with severity of HbE/β0‐thalassaemia. Clinical genetics. 2007; 72:497-505.
- Olivieri NF, Pakbaz Z, Vichinsky E. HbE/β-thalassaemia: basis of marked clinical diversity. Hematology/Oncology Clinics. 2010; 24:1055-70.
- Olivieri NF, Pakbaz Z, Vichinsky E. Hb E/beta-thalassaemia: a common & clinically diverse disorder. The Indian journal of medical research. 2011; 134:522.
- Galanello R, Origa R. Beta-thalassaemia. Orphanet journal of rare diseases. 2010; 5:11.
- Vichinsky E. Haemoglobin E syndromes. ASH Education Program Book. 2007; 2007:79-83.
- George E. HbE β-thalassaemia in Malaysia: revisited. J Hematol Thromb Dis. 2013; 1:2.
- Weatherall DJ & Clegg JB. Inherited haemoglobin disorders: an increasing global health problem. Bull. World Health Organ. 2001; 79:704–12.
- World Health Organization. 1989. Guidelines for the control of haemoglobin disorders. Report of the VIth Annual Meeting of the WHO Working Group on Haemoglobinopathies. World Health Organization, Geneva. Cagliairi, Sardinia.
- Vichinsky EP. Report of Proceedings: 1999 International Conference on E-β Thalassaemia. J. Pediatr. Hematol. Oncol. 2000; 22: 550.
- Marengo-Rowe AJ. The thalassaemias and related disorders. Baylor university medical center proceedings: Taylor & Francis 2007; 27-31.
- Olivieri NF, Muraca GM, O’Donnell A, Premawardhena A, Fisher C, Weatherall DJ. Studies in haemoglobin E beta‐thalassaemia. British journal of haematology. 2008; 141:388-97.
- Abolghasemi H, Amid A, Zeinali S, Radfar MH, Eshghi P, Rahiminejad MS, Ehsani MA, Najmabadi H, Akbari MT, Afrasiabi A, Akhavan-Niaki H. Thalassaemia in Iran: epidemiology, prevention, and management. Journal of Pediatric Hematology/Oncology. 2007; 29:233-38.
- Galanello R, Eleftheriou A, Traeger-Synodinos J, et al. Prevention of Thalassaemias and Other Haemoglobin Disorders. Cyprus: Thalassaemia International Federation Publications.2003.
- Angastiniotis M, Modell B, Englezos P, et al. Prevention and control of haemoglobinopathies. Bull World Health Organ. 1995;73:375–86.
- Tahura S, Selimuzzaman M, Khan WA. Thalassaemia Prevention: Bangladesh Perspective-A Current Update. Bangladesh Journal of Child Health. 2016;40:31-38.
- Weatherall D, Akinyanju O, Fucharoen S, et al. Inherited disordersof Haemoglobin. In: Jamison DT, Breman JG, Measham AR, et al.eds. Disease Control Priorities in Developing Countries. New York:Oxford University Press; 2006:663–80.
- Guidelines for the Management of Non-transfusion dependent thalassaemia (NTDT). 2nd edition. Thalassaemia International Federation.2017; 22.
- Organization WH, et al. Management of haemoglobin disorders: report of a joint WHO-TIF meeting, Nicosia, Cyprus, 16-18 November 2007. 2008.
- Mandal PK, Maji SK, Dolai TK. Present scenario of Haemoglobinopathies in West Bengal, India: An analysis of a large population. Int J Med Public Heal. 2014; 4:496-99.
- Jiang J, Best S, Menzel S et al. cMYB is involved in the regulation of fetal haemoglobin production in adults. Blood.2006; 108:3:1077–83.
- Garner C, Tatu T, Reittie JE et al. Genetic influences on F cells and other hematologic variables: a twin heritability study. Blood.2000; 95:342–46.
- Weatherall D. The thalassaemias. In: Stamatoyannopoulos G, Nienhuis A, Majerus P, eds. Molecular Basis of Blood Diseases (2nd ed). Philadelphia, PA: Saunders; 1994:157.
- Rund D, Rachmilewitz E. Beta-thalassaemia. N Engl J Med. 2005; 353:1135-46.
- Schwartz E Jr. Thalassaemia syndromes. In: Miller D, Baehner R, eds. Smith’s Blood Diseases of Infancy and Childhood (6th ed). St Louis, MO: Mosby; 1989:428.
- Musallam KM, Rivella S, Vichinsky E, Rachmilewitz EA. Non-transfusion-dependent thalassaemias. Haematologica 2013;98:833-44.
- Orkin SH, Kazazian HH Jr, Antonarakis SE, et al. Abnormal RNA processing due to the exon mutation of beta E-globin gene. Nature 1982;300:768–69.
- Datta P, Basu S, Chakravarty SB, et al. Enhanced oxidative cross-linking of Haemoglobin E with spectrin and loss of erythrocyte membrane asymmetry in Haemoglobin Ebeta-thalassaemia. Blood Cells Mol Dis 2006;37:77–81.
- Pootrakul P, Sirankapracha P, Hemsorach S, et al. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in Thai patients with thalassaemia. Blood 2000;96:2606–12.
- Deisseroth, A. et al.Localization of the human alpha-globin structural gene to chromosome 16 in somatic cell hybrids by molecular hybridization assay. Cell.1977;12:205–18.
- Mettananda, S, Gibbons RJ & Higgs, DR. Understanding α-globin gene regulation and implications for the treatment of β-thalassaemia. Annals of the New York Academy of Sciences. 2016; 1368:16–24.
- Higgs DR. The molecular basis of α-thalassaemia. Cold Spring Harbor perpectives in medicine.2013: a011718.
- Deisseroth A et al. Chromosomal localization of human β globin gene on human chromosome 11 in somatic cell hybrids. Proceedings of the National Academy of Sciences of the United States of America.1978; 75:1456–60.
- Noordermeer D and de Laat W. Joining the loops: β-Globin gene regulation. IUBMB Life.2008; 60:824–33.
- Kukreti S et al. Structural polymorphism at LCR and its role in beta globin gene regulation. Biochimie. 2010; 92:1199–206.
- Thein SL and Menzel S. Discovering the genetics underlying foetal haemoglobin production in adults. British Journal of Haematology.2009. 145:455–67.
- Stamatoyannopoulos G. Control of globin gene expression during development and erythroid differentiation. Experimental Hematology.2005; 33: 259–71.
- Boyer SH, Belding TK, Margolet L, Noyes AN: Fetal haemoglobin restriction to a few erythrocytes (F cells) in normal human adults. Science 188:361, 1975.
- Thein SL, Craig JE: Genetics of Hb F/F cell variance in adults and heterocellular hereditary persistence of fetal Haemoglobin. Haemoglobin 1998;22:401.
- Rees DC, Porter JB, Clegg JB, Weatherall DJ. Why are Haemoglobin F levels increased in HbE/β thalassaemia? Blood. 1999; 94:3199-204.
- Forget B: Molecular basis of hereditary persistence of fetal Haemoglobin. Ann N Y Acad Sci.1998;850:38.
- Weatherall DJ, Clegg JB. The Thalassaemia Syndromes. Oxford,UK, Blackwell, 1981.
- Weatherall DJ, Clegg JB, Wood WG, Callender ST, Sheridan BL,Pritchard J: Foetal erythropoiesis in human leukaemia. Nature. 1975; 257:710.
- Blau CA, Constantoulakis P, Al-Khatti A, Spadaccino E, Goldwasser E, Papayannopoulou T, Stamatoyannopoulos G: Fetal Haemoglobin in acute and chronic states of erythroid expansion. Blood. 1993; 81:227.
- Stamatoyannopoulos G, Veith R, Galanello R, PapayannopoulouT: HbF production in stressed erythropoiesis: Observations and kineticmodels. Ann N Y Acad Sci.1985; 445:188.
- Garner C, Tatu T, Game L, Cardon LR, Spector TD, Farrall M, et al. A candidate gene study of F cell levels in sibling pairs using a joint linkage and association analysis. GeneScreen 2000; 1:9-14.
- Badens C, Joly P, Agouti I, Thuret I, Gonnet K, Fattoum S et al. Variants in genetic modifiers of b-thalassaemiacan help to predict the major or intermedia type of thedisease. Haematologica. 2011; 96:1712-14.
- Thein SL, Wainscoat JS, Sampietro M,Old JM, Cappellini D, Fiorelli G et al. Association of thalassaemia intermedia with a β-globin gene haplotype. Br J Haematol 1987;65:367-73.
- Labie D, Pagnier J, Lapoumeroulie C, Rouabhi F, Dunda-Belkhodja O, Chardin P, et al. Common haplotype dependency of high G g-globin gene expression and high Hb F levels in β-thalassaemia and sickle cell anemia patients. Proc Natl Acad Sci USA 1985;82:2111-14.
- Bank A. Regulation of human fetal Haemoglobin: new players, new complexities. Blood. 2006; 10:435-43.
- Schechter A. Haemoglobin research and the origins of molecular medicine. Blood. 2008; 112:3927-38.
- Peri KG, Gagnon J, Gagnon C et al. Association of 158(C>T) (Xmn1) DNA polymorphism in G [gamma]-globin promoter with delayed switchover from fetal to adult haemoglobin synthesis. Pediatr Res.1997; 41:214–17.
- Ghanem M, Girodon E, Vidaud M et al. A comprehensive scanning method for rapid detection of beta-globin gene mutation and polymorphisms. Hum Mutat.1992;1:229-39.
- Gilman JG, Huisman THJ. DNA sequence variation associated with elevated fetal Gγ-globin production. Blood.1985; 66:783–87.
- Garner C, Tatu T, Best S, Creary L, Thein S. Evidence of genetic interaction between the b-globin complex and chromosome 8q in the expression of fetal Haemoglobin. Am J Hum Genet.2002; 70:793–99.
- Hooshang N, Zohreh R, Gholamreza B. The Xmn1 polymorphic site 5’ to the Gγ-gene and its correlation to the Gγ: Aγ ratio, age at first blood transfusion and clinical features in β-thalassaemia patients from Western Iran. Mol Biol Rep. 2010; 37:159-64.
- Najmabadi H, Karimi-Nejad R, Sahebjam S, Pourfarzad F, Teimourian S, Sahebjam F, Amirizadeh N, Karimi-Nejad MH. The β-thalassaemia mutation spectrum in the Iranian population. Haemoglobin.2001;25:285–96.
- Steinberg MH, Forget BG, Higgs DR, Nagel RL (eds). Disorders of Haemoglobin: genetics, pathophysiology, and clinical management. Cambridge University press, Cambridge.2001;356–88.
- Ballas SK, Talacki CA, Adachi K, SchwartzE, Surrey S, Rappaport E. The Xmn1site (-158, C>T) 5’ to the G-gamma gene: correlation with the Senegalese haplotype and G-gamma globin expression. Haemoglobin.1991;15:393–405.
- Nuntakarn L, Fucharoen S, Fucharoen G, et al. Molecular, hematological and clinical aspects of thalassaemia major and thalassaemia intermedia associated with HbE/β-thalassaemia in Northeast Thailand. Blood Cells, Molecules, and Diseases. 2009;42:32–35.
- Adnan RA, Hanafi NS, Alwi Z, Sulong S. Detection of XMN1-Gγ Polymorphism Among Patients with HbE/Βeta Thalassaemia in North East Malaysia. Journal of Biomedical and Clinical Sciences (JBCS). 2018; 2:42-43.
- Irshad S, Muhammad A, Muazzam A, Anmol FS, Shahzad R. Xmn1 Polymorphism: A Silver Lining for β-Thalassaemia Patients. Pakistan Journal of Zoology. 2019; 51:295-300.
- Said F, Abdel-Salam A. XmnI polymorphism: Relation to β-thalassaemia phenotype and genotype in Egyptian Children. Egyptian Journal of Medical Human Genetics. 2015; 16:123-27.
- Oberoi S, Das R, Panigrahi I, Kaur J, Marwaha RK. Xmn1‐Gγ polymorphism and clinical predictors of severity of disease in β‐thalassaemia intermedia. Pediatric blood & cancer. 2011; 57:1025-28.
- Motovali-Bashi M, Ghasemi T. Role of XmnIG Polymorphism in Hydroxyurea Treatment and Fetal Haemoglobin Level at Isfahanian Intermediate β-Thalassaemia Patients. Iranian Biomedical Journal. 2015; 19:177.
- Mokhtar GM, Tantawy AA, Adly AA, ismail EA. Clinicopathological and radiological Study of Egyptian b-Thalassaemia intermedia and ?-Thalassaemia Major patients: relation to Complications and response to Therapy. Haemoglobin 2011; 35: 382-405.
- Ehsani MA, hedayati-Asl AA, Bagheri A, Zeinali S, rashidi A. Hydroxyurea induced hematological response in transfusion-independent beta-thalassaemia intermedia: case series and review of literature. pediatr hematol Oncol.2009; 26: 560-65.
- Ansari SH, Shamsi TS, Ashraf M, Perveen K, Farzana T, Bohray M, Erum S, Mehboob T. Efficacy of hydroxyurea in providing transfusion independence in b thalassaemia. J Pediatr Hematol Oncol 2011; 33: 339-43.
- Zamani F, Shakeri r, Eslami SM, razavi SM, Basi A. hydroxyurea therapy in 49 patients with major beta-thalassaemia. Arch Iran Med. 2009; 12: 295-97.
- Italia KY, Jijina FJ, Merchant R, Panjwani S, Nadkarni AH, Sawant PM, Nair SB, Ghosh K, Colah RB. Response to hydroxyurea in beta-thalassaemia major and intermedia: experience in western india. Clin Chim Acta. 2009; 407:10-15.
- Fucharoen S, Siritanaratkul N, Winichagoon P et al. Hydroxyurea increases haemoglobin F levels and improves the effectiveness of erythropoiesis in beta thalassaemia/haemoglobin E disease. Blood 1996; 87:887-92.
- Kosaryan M, Vahidshahi K, Karami h, Ehteshami S. Effect of hydroxyurea on Thalassaemia Major and Thalassaemia intermedia in iranian patients. Pak J Med Sci 2009; 25:74-78.
- Nadkarni A, Gorakshakar AC, Lu CY, Krishnamoorthy R, Ghosh K, Colah R, Mohanty D. Molecular pathogenesis and clinical variability of beta-thalassaemia syndromes among Indians. Am J Hematol. 2001; 68:75–80.
- Bandyopadhyay S, Roychowdhury K, Chandra S, Das M, Dasgupta UB. Variable severity of β-thalassaemia patients of Eastern India: Effect of α-thalassaemia and XmnI polymorphism. Clinical and experimental medicine. 2001; 1:155-59.
- Kumar R, Kaur A, Agarwal S. Influence of Xmn 1 G γ (HBG2 c.-211 C→ T) Globin Gene Polymorphism on Phenotype of Thalassaemia Patients of North India. Indian Journal of Hematology and Blood Transfusion. 2014; 30:286-90.
- Karimi M, Yarmohammadi H, Farjadian S, Mighaddam Z, cappeliini MD, Giordano PC. Beta thalassaemia intermedia from southern Iran: IVSII.1(G:A) is the prevalent thalassaemia intermedia allele. Haemoglobin. 2002; 26:147–54.
- Wong YC, George E, Tan KL, Yap SF, Chan LL, Tan MA. Molecular characterisation and frequency of Gγ Xmn I polymorphism in Chinese and Malay β thalassaemia patients in Malaysia. Malaysian Journal of Pathology. 2006; 28:17-21.